

अपने करियर के पिछले दशकों में, मैंने दवाओं की सुरक्षा पर शोध करके अमेरिकियों की सुरक्षा के लिए अनगिनत घंटे बिताए हैं। मेरी शिक्षा और करियर ने मुझे लगभग आधा दर्जन विश्वविद्यालयों, बिग फार्मा और तीन राष्ट्रपति प्रशासनों के तहत एफडीए में पहुंचाया है। दवा सुरक्षा इस बात पर विचार करती है कि क्यों एक व्यक्ति एक फार्मास्युटिकल उत्पाद ले सकता है और उस पर कोई प्रतिकूल प्रभाव नहीं पड़ता है, जबकि एक अलग व्यक्ति एक ही उत्पाद ले सकता है लेकिन उसे स्थायी विकलांगता या मृत्यु तक की प्रतिकूल प्रतिक्रिया हो सकती है। डिफ़ॉल्ट रूप से, दवा सुरक्षा का अध्ययन विनिर्माण और दवा की गुणवत्ता के गैर-नैदानिक पहलुओं पर भी विचार करता है।
क्योंकि दवा की सुरक्षा का आकलन करने के लिए दवा की गुणवत्ता एक आवश्यक कारक है, अमेरिकियों की सुरक्षा के लिए मेरे प्रयास ने मुझे दुनिया के पहले "की अवधारणा और स्थापना के लिए प्रेरित किया।"विश्लेषणात्मक फार्मेसी“भारत और चीन जैसे स्थानों से फार्मास्युटिकल उत्पादों को रोगियों को वितरित करने से पहले वैज्ञानिक रूप से सत्यापित करने का मिशन। दुर्भाग्य से, नैतिकता से ऊपर उदारता की खोज और मरीजों की सुरक्षा के कारण उस कंपनी को वित्तीय प्रबंधन करना पड़ा व्यापक एफडीए उल्लंघन और न्यायाधीशों द्वारा बनाने का आरोप लगाया जा रहा है झूठे वैज्ञानिक दावे (यह सब मेरे बाहर निकलने के बाद संयोगवश घटित हुआ)।
दवा की गुणवत्ता की बाहरी पुष्टि के बिना, अमेरिकी उत्पाद की शुद्धता का आकलन और पुष्टि करने के लिए पूरी तरह से एफडीए और निर्माताओं पर निर्भर हैं। जब कोविड एमआरएनए इंजेक्शन की बात आती है तो दवा सुरक्षा को एक उल्लेखनीय समस्या के रूप में देखा गया है। दुर्भाग्य से, यदि कोई एमआरएनए इंजेक्शन पर अपना स्वयं का विश्लेषण करना चाहता है, तो वे इसकी तुलना करने के लिए उचित रूप से विस्तृत घटक सूची नहीं है, या शुद्धता के लिए इसकी उचित जांच कैसे करें, इस पर स्थापित नियामक पद्धति तक पहुंच भी नहीं है.
यह निर्माताओं के कारण है और एफडीए इन एमआरएनए इंजेक्शनों के सभी अवयवों पर विचार करता है, जिसमें एमआरएनए प्लस लिपिड नैनोपार्टिकल (एलएनपी) गुणों का अनुक्रम शामिल है, जिसमें आधा जीवन, एलएनपी संरचनाएं, सतह संशोधन, प्रति खुराक एलएनपी की संख्या/प्रकार और अनुलग्नक बिंदु शामिल हैं। एमआरएनए स्ट्रैंड, अनिर्दिष्ट या "व्यापार रहस्य" होना।
इसके अलावा, एफडीए अतिरिक्त रूप से विचार करता है के तरीके शुद्धता के लिए एमआरएनए इंजेक्शन का परीक्षण कैसे करें, यह भी एक व्यापार रहस्य है।
द्विदलीय समर्थन और करोड़ों करदाता डॉलर, लेकिन कोई पारदर्शिता नहीं?
कोविड एमआरएनए गोपनीयता मौजूद है, भले ही ट्रम्प और बिडेन दोनों प्रशासनों ने कोविड एमआरएनए बौद्धिक संपदा अधिकारों को हटाने के बिंदु पर एमआरएनए इंजेक्शन के साथ पूर्ण पारदर्शिता का प्रस्ताव दिया था। इसके बावजूद, एफडीए और निर्माता दोनों एक व्यापार रहस्य के रूप में इन शॉट्स के बारे में बुनियादी डेटा सहित पेटेंट पर सख्त पकड़ रखने की अनुमति दे रहे हैं। सभी कोविड वैक्सीन निर्माताओं को मिलने के बावजूद वे ऐसा कर रहे हैं करोड़ों करदाताओं का डॉलर के अनुसार फोर्ब्स/स्टेटिस्टा प्रकाशनों।
औषधि सुरक्षा महामारी विज्ञान का अध्ययन करना काफी कठिन है। सत्यापन योग्य उत्पाद शुद्धता/संगति के बिना, पूर्ण सुरक्षा मूल्यांकन असंभव है।
सभी सामग्रियों की पूर्ण पारदर्शिता और गुणवत्ता नियंत्रण उपाय न केवल इसलिए महत्वपूर्ण हैं क्योंकि उन्हें करदाताओं द्वारा सैकड़ों मिलियन डॉलर से वित्त पोषित किया गया था, बल्कि इसलिए भी क्योंकि कोविड एमआरएनए इंजेक्शन की सुरक्षा और प्रभावकारिता के बारे में कई सवाल उठे हैं।
असाधारण रूप से जटिल होने के अलावा, इसके बाद नियामकों द्वारा उनकी मंजूरी में तेजी लाई गई एक वर्ष से कम. अधिकांश दवाएं और टीके आम तौर पर लगते हैं दस साल सुरक्षा/प्रभावकारिता के लिए पूरी तरह से परीक्षण करना और समीक्षा करना और अनुमोदन करना। सामग्री पूरी तरह से नवीन, बहुत जटिल और बड़े पैमाने पर प्रशासित होने वाली अपनी तरह की पहली होने के अलावा, इसमें विकास भी शामिल है दीर्घकालिक नैदानिक सुरक्षा/विषाक्तता मूल्यांकन और महामारी विज्ञान समीक्षा में तेजी लाई गई और संभवतः रिलीज से पहले पूरी तरह से स्पष्ट नहीं किया गया।
एफडीए संघटक सत्यापन, पारदर्शिता और "सच्चाई" की मिसालें 1800 के दशक की हैं:
विश्लेषणात्मक सत्यापन और सामग्री की पारदर्शिता या "लेबलिंग में सच्चाई" जहां बोतल की सामग्री है अपेक्षित सूचीबद्ध सामग्रियों से मिलान करने के लिए एफडीए की स्थापना 1862 से भी पहले की है. आज का एफडीए वास्तव में अमेरिकी कृषि विभाग में कार्यरत "रसायन विज्ञान विभाग" के एक कर्मचारी के रूप में शुरू होने से पैदा हुआ था।
मिलावट, (परिवर्तित या विषाक्त सामग्री) गलत नामकरण (इसमें गलत लेबल है या अन्यथा भ्रामक है, या इसमें गलत चिकित्सा दावे शामिल हैं), या गलत लेबलिंग (इसमें शामिल घटक उत्पाद लेबल पर सूचीबद्ध नहीं हैं) इन सभी का अमेरिका में लंबा, बदसूरत इतिहास रहा है। ऐसा माना जाता था कि 19वीं सदी की शुरुआत से लेकर मध्य तक उग्रता चरम पर थी - या कम से कम तब यह पहचान योग्य हो गई थी - क्योंकि 1862 तक ही घटक धोखाधड़ी का विश्लेषण और पता लगाने के लिए तकनीकी प्रक्रियाएं विकसित की गई थीं। इससे पहले, तथाकथित "यात्रा करने वाले चिकित्सक" जो खुद को "डॉक्टर" कहते थे (निश्चित रूप से संदिग्ध या अस्तित्वहीन साख के साथ) "सभी का इलाज करने वाले" उत्पादों की बोतलें बेचते थे, जिनके घटक लेबल केवल अस्पष्ट या अहानिकर सामग्री को सूचीबद्ध करते थे जैसे कि “विटामिन""हर्बल अर्क,या "सर्प तेल”- या अक्सर इसमें कोई घटक सूची ही नहीं होती।
उस समय, कई धर्मनिष्ठ, शुद्धतावादी न्यू इंग्लैंडवासी, जो धार्मिक कारणों से ऐसा करते थे कभी नहीँ शराब को छूते हैं, तो इन तस्करों से इन समाधानों को खरीदेंगे और अनजाने में उन समाधानों का सेवन करने के लिए धोखा खाएंगे जिनमें न केवल शराब होती है, बल्कि अफ़ीम और/या कोकीन जैसे नशीले पदार्थ भी होते हैं। बीमारियों की बेतुकी व्यापक श्रेणी में सुधार करने के बहाने, रोगियों ने इसके बजाय दंडात्मक लत विकसित की और/या अन्यथा उनके स्वास्थ्य पर इन शुरुआती "ड्रग डीलरों" द्वारा नकारात्मक प्रभाव पड़ा।
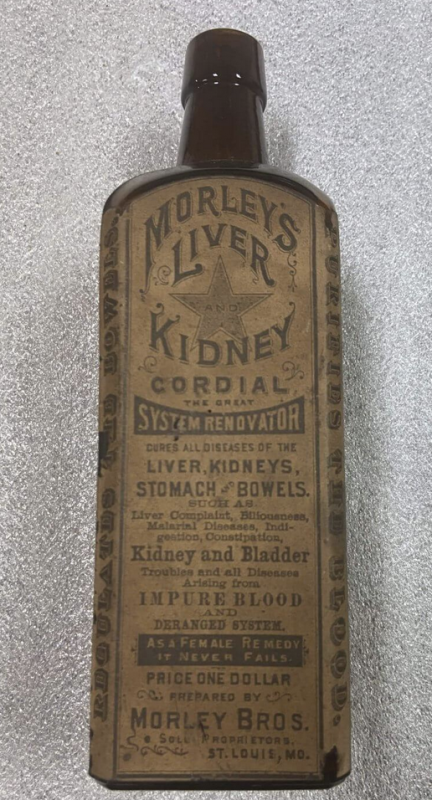
जैसे-जैसे समस्या बढ़ती गई, संघीय सरकार ने इस पर ध्यान देना शुरू कर दिया। अंततः, शुद्ध खाद्य और औषधि अधिनियम 1906 में पारित किया गया और खाद्य एवं औषधि प्रशासन (एफडीए) का निर्माण हुआ।
[एफडीए के पास था रचनात्मक यह सुनिश्चित करने का कर्तव्य है कि दवाएं सत्य लेबलिंग विवरण रखती हैं और शुद्धता और ताकत के लिए कुछ मानकों को पूरा करती हैं।
याद कीजिए करीब 120 साल पुराना सच्ची लेबलिंग आवश्यकता और 1906 के शुद्ध खाद्य एवं औषधि अधिनियम का "शुद्धता" भाग, जैसा कि आप एमआरएनए सत्यापन परीक्षण और घटक पारदर्शिता के बारे में पढ़ते हैं।]
एफडीए-विनियमित उत्पादों के लिए कौन सा "सच्चा" और "शुद्ध" घटक सत्यापन परीक्षण हो रहा है?
2021 में, FDA ने एक के माध्यम से अमेरिका की दवा गुणवत्ता की निगरानी शुरू करने का विकल्प चुना दूरस्थ संग्रह of नमूनों को मेल द्वारा प्रस्तुत करना कोविड महामारी के कारण लाइव सुविधा निरीक्षण के विकल्प के रूप में दवाओं के लिए। क्या वह कानूनी था? क्या इसे कभी वैज्ञानिक दृष्टि से उचित माना जा सकता है? आज, महामारी समाप्त होने के बावजूद, वर्तमान में एकमात्र आधिकारिक फार्मास्युटिकल रिलीज़ परीक्षण किया जा रहा है कोई कोविड एमआरएनए फार्मास्युटिकल प्रकट होता है सेवा मेरे अभी भी एफडीए द्वारा निर्माता-आपूर्ति के माध्यम से किया जाना चाहिए, "मेल-इन" एक के अनुसार नमूना वर्तमान FDA वेबसाइट का स्क्रीनशॉट. जाहिर है, "मेल-इन" नमूनाकरण विधि प्रत्यक्ष, व्यक्तिगत संग्रह विधि के माध्यम से सीधे नमूने एकत्र करने की तुलना में कहीं अलग और संभावित रूप से कम विश्वसनीय है। इसके बावजूद, एफडीए का दावा है कि उसने "नमूनाकरण और परीक्षण के लिए दुनिया भर में उच्चतम मानक".
इसके अलावा, एफडीए अपनी "मेल-इन" दूरस्थ परीक्षण नीति को और आगे बढ़ाने का प्रस्ताव कर रहा है नव प्रस्तावित मार्गदर्शन दस्तावेज़.
हालाँकि यह केवल "ड्राफ्ट" FDA दस्तावेज़ के रूप में मौजूद है, आधिकारिक FDA वेबसाइटें इसे दिखाती हैं ऐसा प्रतीत होता है कि नमूनों की मेलिंग कम से कम जनवरी 2021 से पहले ही लागू हो चुकी है. ऐसा प्रतीत होता है कि एफडीए उन मेल-इन परीक्षणों के परिणामों को उनके स्वतंत्र सत्यापन के रूप में दावा कर रहा है।
इसके अलावा, FDA ड्राफ्ट के पहले पृष्ठ के नीचे दस्तावेज़ "दूरस्थ परीक्षण" के विस्तार का प्रस्ताव करता है। यह वर्तमान में सूचीबद्ध है प्रत्येक एफडीए में एफडीए उत्पाद-विनियमन प्रभाग, जिसका अर्थ है कि यह एक एजेंसीव्यापी नीति प्रस्ताव है।
पूरी सूची में ये शामिल हैं:
- नियामक मामलों का कार्यालय
- खाद्य नीति और प्रतिक्रिया कार्यालय
- संयोजन उत्पादों का कार्यालय
- बायोलॉजिक्स मूल्यांकन और अनुसंधान केंद्र
- दवा मूल्यांकन और अनुसंधान के लिए केंद्र
- उपकरण और रेडियोलॉजिकल स्वास्थ्य केंद्र
- खाद्य सुरक्षा और अनुप्रयुक्त पोषण केंद्र
- तम्बाकू उत्पाद केन्द्र
- पशु चिकित्सा केंद्र
क्या एफडीए द्वारा "मेल द्वारा भेजा गया" गुणवत्ता नियंत्रण नमूनाकरण उपयुक्त है? क्या होगा यदि राज्यों के स्वास्थ्य विभाग के रेस्तरां निरीक्षण एफडीए नीति को प्रतिबिंबित करते हैं?
यह "मेल-इन" नमूनाकरण पद्धति भी इसी तरह बेतुकी है, उदाहरण के लिए, राज्यों के स्वास्थ्य विभाग द्वारा रेस्तरां की निगरानी करने के लिए उन्हें समय-समय पर अपने मेनू से विभिन्न वस्तुओं को परीक्षण सुविधा में "मेल" करने के लिए कहा जाता है ताकि स्वास्थ्य विभाग संभावित भोजन के लिए परीक्षण कर सकें। -जन्मजात संदूषण, और/या रेस्तरां से मेनू आइटमों का स्वयं परीक्षण करने का वादा करने के लिए कहना। यदि वह रेस्तरां चीन में होता तो क्या होता? अगर वह रेस्तरां भारत में होता तो क्या होता? या कोई अन्य देश जिसके लिए प्रसिद्ध है धोखाधड़ी और गुणवत्ता नियंत्रण का घिनौना इतिहास समस्या का?
वह पद्धति रेस्तरां और फार्मास्युटिकल कंपनियों दोनों के लिए अस्वीकार्य होगी, जिनमें स्पष्ट कारण शामिल हैं: निर्माता अपनी पसंद के नमूने भेज सकते हैं - जरूरी नहीं कि प्रतिनिधि बैच के नमूने हों। यह स्पष्ट रूप से एफडीए निरीक्षकों द्वारा संपूर्ण सुविधा के अघोषित निरीक्षण के दौरान नमूने प्राप्त करने के समान नहीं है।
रेस्तरां सादृश्य के तहत, निश्चित रूप से सभी रेस्तरां ऐसा करेंगे "ए" ग्रेड नमूने जमा करें जो जरूरी नहीं कि उपभोक्ताओं को जो मिलता है उसका प्रतिनिधि हो।

गुणवत्ता नियंत्रण: फार्मास्युटिकल "रिलीज़ परीक्षण" क्या है और यह महत्वपूर्ण क्यों है?
आज, एफडीए गुणवत्ता और सामग्री की देखरेख करता है $2.7 ट्रिलियन सालाना उत्पाद का मूल्य, लेकिन ऐसा लगता है कि यह महत्वपूर्ण घटक सत्यापन आकलन और परिणामों को दबा रहा है। एफडीए का उद्देश्य आचरण करके अमेरिकियों की रक्षा करना है व्यापक घटक सटीकता सुनिश्चित करने के लिए चेकसम के रूप में विश्लेषणात्मक परीक्षण। इसके परिणाम उन करदाताओं के लिए पारदर्शी होने चाहिए जो फंड देते हैं FDA का $6.6 बिलियन बजट। उस वैज्ञानिक सत्यापन को फार्मास्युटिकल कहा जाता है"परीक्षण जारी करें।” रिलीज़ परीक्षण एक तकनीकी शब्द है जो एक ऐसी प्रक्रिया को संदर्भित करता है जिसमें विभिन्न प्रकार के उपकरणीय विश्लेषण शामिल होते हैं व्यापक शुद्धता, एकाग्रता, स्थिरता, पहचान और किसी भी प्रकार की अशुद्धियों के लिए उत्पादों का परीक्षण करें।
पूरे एफडीए का जन्म 1862 में उस एक "रसायन विज्ञान विभाग" कर्मचारी से हुआ था और सामग्री की पारदर्शिता और सत्यापन की आवश्यकता थी। आज, वह कर्मचारी एक के रूप में विकसित हो गया है 1,300 वैज्ञानिकों और सहायक कर्मचारियों का संपूर्ण FDA विभाग फार्मास्युटिकल रिलीज परीक्षण के माध्यम से घटक सत्यापन के लिए अनुमानित रूप से समर्पित। एफडीए का औषधि गुणवत्ता कार्यालय (ओपीक्यू) को यह सुनिश्चित करना चाहिए कि फार्मास्यूटिकल्स गुणवत्ता/अशुद्धता (गुणात्मक) या सामग्री (गुणात्मक) परिवर्तनशीलता के बिना, सूचीबद्ध सामग्रियों की सामग्री से बिल्कुल मेल खाते हैं। इसके लिए आवश्यक नियम बहुत विशिष्ट और विस्तृत हैं 21 CFR N 201.10.
एफडीए गुणवत्ता नियंत्रण के लिए एमआरएनए इंजेक्शन का सत्यापन कैसे करता है:
एमआरएनए इंजेक्शन से परीक्षणों से गुणवत्ता नियंत्रण परिणाम विशेष रूप से महत्वपूर्ण थे क्योंकि वे बड़े, जटिल हैं और तेजी से बनाए गए थे। जबकि करदाता एमआरएनए इंजेक्शन की गुणवत्ता की पुष्टि करने और परिणाम साझा करने के लिए एफडीए पर निर्भर हैं लगता है एमआरएनए कोविड उत्पादों के संबंध में सबसे बुनियादी पारदर्शिता की कीमत पर भी निर्माताओं की सामग्री की रक्षा करने के लिए बाध्य है। जबकि एफडीए नमूने एकत्र कर रहा है, उनकी "मेल-इन" पद्धति मौलिक रूप से त्रुटिपूर्ण है। इसके अतिरिक्त, एफडीए उन परीक्षणों के परिणामों को कहीं भी साझा नहीं कर रहा है जहां मैं उन्हें पा सकता हूं।
दूसरे शब्दों में: महामारी के दौरान जब बिल्कुल नए, व्यापक रूप से कार्यान्वित एमआरएनए शॉट्स अमेरिकियों पर "तानाशाही गति" से थोपे जा रहे थे और जब अमेरिका एफडीए की गुणवत्ता/नियामक कर्तव्यों पर सबसे अधिक भरोसा कर रहा था, एफडीए स्व-प्रस्तुत "मेल से" स्वीकार कर रहा था। गुणवत्ता नियंत्रण परीक्षण और/या परिणाम। क्या एफडीए ने इस पर विचार नहीं किया एमआरएनए निर्माताओं ने स्वीकार किया कि वे विनिर्माण के प्रति प्रतिक्रिया देने के लिए "संघर्ष" कर रहे हैं और आगे बढ़ने के लिए "संघर्ष" कर रहे हैं। विनिर्माण प्रक्रियाओं के साथ? एमआरएनए सामग्री के निर्माताओं ने आगे कहा कि जरूरतों को पूरा करने के प्रयास "अभूतपूर्व" थे।
इस तरह के बयान गुणवत्ता में उपभोक्ता का विश्वास पैदा नहीं करते हैं, और इन जटिल उत्पादों की जबरदस्त वृद्धि का उदाहरण हैं जिनकी गारंटी दी जानी चाहिए विशेष रूप से सतर्क और व्यक्तिगत रूप से सुविधाओं और विनिर्मित उत्पादों की एफडीए जांच, चाहे महामारी हो या नहीं। उदाहरण के लिए, एक एमआरएनए घटक निर्माता ने कहा कि उन्होंने अचानक अपना उत्पादन बढ़ा दिया है 50 गुना.
"ताना-बाना" जल्दबाजी में आगे बढ़ाई गई उस नई तकनीक के बीच, एफडीए के 1,300 ओपीक्यू वैज्ञानिकों में से कोई भी लाइव निरीक्षण की मांग नहीं कर रहा था, या कम से कम, संभावित रूप से संदिग्ध "मेल-इन" नमूने मांगने के अलावा कुछ भी करने की पेशकश नहीं कर रहा था। परीक्षण के लिए?
स्पष्ट प्रश्न है: एफडीए ने सीधे नमूने क्यों नहीं एकत्र किए? यहां तक कि महामारी के बावजूद, एफडीए खतरनाक सूट पहनकर या - या पर सुविधाओं का निरीक्षण कर सकता था बहुत कम से कम - फार्मेसियों, अस्पतालों, या वितरक गोदामों से नमूने एकत्र करने का विकल्प चुना।
एमआरएनए इंजेक्शन सामग्री के परीक्षण के लिए गुप्त पद्धति:
परीक्षण परिणामों की अनुपस्थिति और संदिग्ध "मेल-इन" नमूनाकरण परिणामों से परे - एफडीए है के अतिरिक्त अपनी मान्य कार्यप्रणाली को छिपाकर दूसरों को एमआरएनए इंजेक्शन की गुणवत्ता/शुद्धता पर अपना स्वयं का स्वतंत्र विश्लेषण करने से रोकना।
घटक सूची की तुलना में शुद्धता और संभावित संदूषण के लिए दवाओं का स्वतंत्र रूप से विश्लेषण करना कुछ ऐसा है जिसे मैंने खुद करने का प्रयास किया था जब मैंने दुनिया की पहली अवधारणा तैयार की थी। विश्लेषणात्मक फार्मेसी. हालाँकि, चूंकि एमआरएनए शॉट्स पूरी तरह से कम-पारदर्शी घटक सूची के साथ एक नई तकनीक है, इसलिए जिस परीक्षण पद्धति को नियोजित करने की आवश्यकता होगी वह सीधी नहीं है क्योंकि यह अन्य छोटे-अणु दवाओं के लिए होगी। भंडारण, स्थिरता, विशिष्टता, रसायन शास्त्र, संवेदनशीलता, या यहां तक कि परीक्षण सत्यापन और/या परिणामों के लिए बुनियादी पद्धति को देखने की कोशिश करने वाले किसी भी व्यक्ति को एफडीए रिपोर्ट के माध्यम से अवरुद्ध कर दिया जाता है जिसमें हास्यास्पद आक्रामक कटौती होती है, यहां तक कि संभावित मूल्यांकन करने के तरीके की सबसे मौलिक वैज्ञानिक समझ भी बन जाती है। परिणाम या परीक्षण करना असंभव है।
एक मार्मिक दृश्य उदाहरण के रूप में, एक लंबे एफडीए विनियामक सारांश (नीचे दिखाया गया है) में एक संपादित पृष्ठ एक का हिस्सा है 127 पृष्ठों के इस दस्तावेज (जिनमें से केवल 63 पृष्ठ साझा किए गए हैं, और उन 63 पृष्ठों में से लगभग 50% को संशोधित किया गया है) एमआरएनए इंजेक्शन की शुद्धता, एकाग्रता और अन्य विश्लेषणात्मक उपायों का मूल्यांकन कैसे करें।
उन एफडीए (बी)(4) कटौती निर्दिष्ट विस्तृत संशोधनों का उपयोग "व्यापार रहस्यों और गोपनीय वाणिज्यिक या वित्तीय जानकारी की रक्षा करना।” लेकिन यदि अनुसंधान/विकास/उत्पाद को वित्त पोषित किया गया था तो क्या इसे "वाणिज्यिक" लेबल करना वास्तव में उचित है करोड़ों करदाताओं का डॉलर?

अवयवों की सूची या परीक्षण पद्धति के बिना, एफडीए या निर्माताओं के बाहर किसी अन्य व्यक्ति के लिए यह जानना असंभव है कि उत्पाद की जांच कैसे करें मिलावट (परिवर्तित या विषैले तत्व) या गलत लेबलिंग (क्योंकि न्यूक्लियोटाइड अनुक्रम और सहित घटक(ओं) की पूरी सूची लिपिड नैनोकण विन्यास उत्पाद लेबल पर विशेष रूप से अस्पष्ट हैं)।
कार्यप्रणाली की कमी विशेष रूप से परेशानी वाली है क्योंकि स्वतंत्र पद्धति का उपयोग करने वाले नए, प्रारंभिक डेटा ने इसके प्रमाण दिखाए हैं एमआरएनए कोविड इंजेक्शन में डीएनए संदूषण.
इसलिए, यदि कोई बाहरी व्यक्ति परीक्षण करने का दावा करता है और एमआरएनए शॉट्स में अशुद्धता पाता है और एफडीए या निर्माताओं से इसकी प्रतिक्रिया मांगता है, तो उन्हें कुछ इस तरह की प्रतिक्रिया दी जाएगी:
- आपने अपने निष्कर्ष पर पहुंचने के लिए मान्य/उपयुक्त परीक्षण पद्धति का उपयोग नहीं किया और इसलिए आपके विश्लेषण अमान्य हैं।
इसके लिए, स्वतंत्र प्रयोगशाला एफडीए-अनुमोदित दस्तावेज़ (यानी, पूर्ण दस्तावेज़ युक्त) से परीक्षण पद्धति का अनुरोध करने का प्रयास करेगी आकृति 5) पूछकर: “ठीक है, मैं आपकी अनुमोदित पद्धति का उपयोग करके इसका परीक्षण करना चाहूंगा; क्या आप हमें बताएंगे कि वह क्या है?”
- एफडीए या निर्माता कुछ इस प्रकार उत्तर देंगे: "हम अपनाई गई कार्यप्रणाली के बारे में जो खुलासा करना चाहते हैं वह गोपनीय नहीं है, वह ऑनलाइन या एफडीए एफओआईए अनुरोध के माध्यम से पाया जा सकता है।”…जहां उनसे मुलाकात होगी निम्नलिखित भारी संशोधित दस्तावेज़, जहां दूर से अर्थपूर्ण कोई भी चीज़ (बी)(4) संशोधनों से ढकी हुई है।
पंक्तियों के बीच में पढ़ना: यह स्पष्ट है कि निर्माता और अमेरिका का एफडीए दोनों नहीं चाहते हैं कि उनके अलावा कोई और व्यक्ति एमआरएनए इंजेक्शन की पूरी सामग्री को जाने या यहां तक कि शुद्धता और स्थिरता के लिए एमआरएनए इंजेक्शन का परीक्षण भी करे।
एफडीए अधिकारियों के मुताबिक: फार्मास्युटिकल मैन्युफैक्चरिंग है अत्यधिक त्रुटि प्रवण:
बहुत फार्मास्युटिकल निर्माण प्रक्रिया के दौरान चीजें गलत हो सकती हैं - और होती भी हैं। एमआरएनए/एलएनपी इंजेक्शन के साथ संभावित विसंगतियों से परे, गुणात्मक और मात्रात्मक मुद्दे शामिल हैं प्रत्येक एफडीए-विनियमित फार्मास्युटिकल उत्पाद। यहां तक कि सदन और सीनेट ने भी औपचारिक रूप से अमेरिका की दवा आपूर्ति श्रृंखला को सुरक्षित करने में एफडीए की विफलता की रिपोर्ट को स्वीकार कर लिया है। अधिकांश अमेरिका की दवा उपभोक्ता-अंत-उपयोगकर्ता उत्पादका उत्पादन किया जा रहा है विदेशों में भारत और चीन जैसे देशों में, और अन्य कम श्रम लागत वाले देश हैं नहीं गुणवत्ता नियंत्रण के उच्च स्तर के लिए जाना जाता है. संघीय रजिस्टर रिपोर्टों से भरा पड़ा है भारतीय और चीनी विनिर्माण संयंत्रों में उल्लंघन.
क्या एफडीए इन संयंत्रों को भी प्रमाणित कर रहा है - जिनमें उल्लंघन के लंबे इतिहास वाले संयंत्र भी शामिल हैं - एफडीए को "मेल-इन" प्रणाली के माध्यम से? अपमानजनक रूप से, प्रश्न का उत्तर कुछ ऐसा है जो दवा की गुणवत्ता से संबंधित किसी भी व्यक्ति को बहुत असहज कर देगा।
थोड़ी देर सिक्स सिग्मा ऑटोमोबाइल, कंप्यूटर, मोबाइल टेलीफोन और अन्य उच्च तकनीक विनिर्माण में गुणवत्ता और सुरक्षा के लिए परिशुद्धता स्तर लंबे समय से लक्ष्य रहा है, ऐसा लगता है कि जब फार्मास्युटिकल विनिर्माण की बात आती है तो इसे ज्यादातर नजरअंदाज कर दिया गया है।
एफडीए अधिकारियों ने फार्मास्युटिकल विनिर्माण में 2-3σ (सिग्मा) की अशुद्धता का अनुमान लगाते हुए डेटा प्रकाशित किया है। एक 2σ गुणवत्ता से मेल खाती है प्रति 308,537 अवसरों पर 1,000,000 दोष. (जब फार्मास्युटिकल निर्माण की बात आती है तो त्रुटि के 1,000,000 से अधिक अवसर होने की संभावना है।) एफडीए नेतृत्व के उच्चतम स्तर पर इसके बारे में जानता है; वास्तव में, वर्तमान एफडीए के फार्मास्युटिकल गुणवत्ता कार्यालय के प्रमुख, माइकल कोप्चा यहां तक कि फार्मास्युटिकल निर्माण की अस्पष्ट प्रकृति पर अफसोस जताते हुए उपरोक्त सिक्स सिग्मा गणना को लिखा और प्रकाशित किया वापस 2017.
एमआरएनए उत्पादों और/या उनके एलएनपी के लिए त्रुटि का अक्षांश सम हो सकता है कम 2-3σ से सटीक, (σ जितना कम होगा, उत्पाद उतना ही अधिक गलत होगा) क्योंकि उनमें न्यूक्लियोटाइड सामग्री और उपन्यास एलएनपी शामिल होते हैं, जो उन्हें छोटे-अणु फार्मास्यूटिकल्स की तुलना में काफी अधिक जटिल बनाते हैं - भले ही उनका विकास, निर्माण और विमोचन किया जा रहा हो। खराब गति।"
यहां तक कि एफडीए और उसके अधिकारी भी अंतर्निहित विनिर्माण अशुद्धि को पहचान रहे हैं, खेल की विस्तृत दुनिया में क्यों क्या एफडीए सार्वजनिक रूप से एमआरएनए प्रौद्योगिकी के अपने रिलीज परीक्षण को अमेरिकी जनता के साथ साझा करके अपने सुरक्षा मिशन को पूरा नहीं कर रहा है जो उन्हें वित्त पोषित करता है?
1862 से पहले फिर से? क्या एमआरएनए शॉट्स ही एकमात्र दवा है जो अमेरिकियों के पास नहीं है? पूर्ण संघटक जानकारी?
एमआरएनए शॉट्स और अन्य महत्वपूर्ण जानकारी के अनुक्रमों की संख्या पर स्पष्टता की कमी एक अन्य एफडीए-अनुमोदित आरएनए-आधारित दवा के सीधे विपरीत है - पतिसिरन (ऑनपैट्रो®). ऑनपैट्रो पारदर्शी रूप से आधिकारिक एफडीए के भीतर अपने उत्पादों का अनुक्रम, आणविक भार और मिलीग्राम शक्ति प्रदान करता है पैकेज लेबलिंग जैसा कि नीचे एक अंश में दर्शाया गया है:


कोविड एमआरएनए खुराक की कमी विशिष्टता: 0.3 एमएल (या 0.5 एमएल) किस?
अभी तक, हमारे पास किसी भी कोविड एमआरएनए इंजेक्शन के बुनियादी घटक की जानकारी नहीं है। फार्मासिस्ट केवल एक विशेष जानकारी देना जानते हैं आयतन तरल पदार्थ का, और ऐसा प्रतीत होता है कि उसने बिना किसी प्रश्न के ऐसा किया। आम तौर पर, आधिकारिक एफडीए पैकेज लेबलिंग में उस मात्रा में वास्तविक सामग्री का विवरण होना चाहिए, लेकिन कोविड एमआरएनए लेबल के लिए नहीं: वे केवल 0.3 एमएल (या 0.5 एमएल) को "खुराक फॉर्म और ताकत" के रूप में बताते हैं।
इसके अतिरिक्त, जैसा कि कोई भी हाई स्कूल का छात्र आपको बता सकता है, 0.3/0.5mL एक है आयतन, नहीं a शक्ति. हमें उस 0.3/0.5 एमएल में क्या शामिल है इसकी कोई मात्रात्मक जानकारी नहीं है जैसे: कितने एलएनपी कण? उन एलएनपी का आकार/आकार क्या है? उस आयतन में कितने mRNA अनुक्रम हैं?




क्या यह एफडीए द्वारा पर्याप्त रूप से पारदर्शी या "सच्ची लेबलिंग" के रूप में प्रमाणित है?
पैकेज इंसर्ट से उपरोक्त कट-एंड-पेस्ट अंश वह सारी जानकारी है जो निर्माता खुराक के संबंध में उपभोक्ताओं के साथ साझा कर रहे हैं - जो कि अन्य सभी एफडीए लेबल की तुलना में बेहद अपर्याप्त है - या कोई भी जो कितना तरल पदार्थ से परे कुछ भी जानने के लिए उत्सुक है इंजेक्ट करने के लिए और एक अनिर्दिष्ट mRNA अनुक्रम की 30 या 100mcg सांद्रता।
एफडीए द्वारा अनुमत इस लेबल की उल्लेखनीय अशुद्धता विशेष रूप से इसके लगभग 120 साल पुराने लेबल के साथ विरोधाभासी प्रतीत होती है: "यह आवश्यक है कि भोजन और औषधियों पर सही लेबलिंग विवरण हो और वे शुद्धता और ताकत के लिए कुछ मानकों को पूरा करें".
क्या यह एफडीए द्वारा सामग्री की "सच्ची" सूची के रूप में मान्य है? (देखना 21सीएफआर §352, तथा 21 सीएफआर 201.10 "सामग्रियों का विवरण" और "गलत ब्रांड वाली दवाओं और उपकरणों" के संबंध में।)
सवाल यह है: क्या अज्ञात या गैर-विशिष्ट सामग्रियों को सूचीबद्ध करना है जिन्हें निर्माता के अलावा कोई भी समझ नहीं सकता है वास्तव में "लेबलिंग?" की भावना या कानूनी आवश्यकताओं को पूरा करें? क्या वह लेबल जिसे अमेरिका का FDA "सच्चा" मानता है? वैसे भी एफडीए किसकी तरफ है; निर्माता या उपभोक्ता?
इसके अलावा इसे सीधे निर्दिष्ट नहीं किया जा सकता है, 30 या 100 एमसीजी इंजेक्शन में एलएनपी और न ही एमआरएनए स्ट्रैंड की सटीक संख्या का अनुमान भी नहीं लगाया जा सकता है। Stoiciometrically या के आधार पर अवोगाद्रो का नंबर, क्योंकि एमआरएनए अनुक्रम, आणविक भार, और/या एलएनपी घटक/कॉन्फ़िगरेशन आधिकारिक एफडीए लेबलिंग के भीतर कहीं भी प्रदान नहीं किए गए हैं।
कोई कैसे जान सकता है कि कोविड के लिए स्पाइक प्रोटीन को एनकोड करने के लिए एमआरएनए स्ट्रैंड की संख्या, समुदाय से प्राप्त संक्रमण से प्राप्त होने वाले कोविड इनोकुलम के भार के समानुपाती है? उत्तर: वे नहीं कर सकते.
क्या कोविड एमआरएनए इंजेक्शन हैं? उचित रूप से लेबल/गलत लेबल किया गया?
21 सीएफआर 211.125 निर्दिष्ट करता है "दवा उत्पाद लेबलिंग कार्यों में उपयोग के लिए जारी लेबलिंग पर सख्त नियंत्रण रखा जाएगा।“लेकिन ऐसा प्रतीत होता है कि एफडीए इस तथ्य के बावजूद कोविड एमआरएनए इंजेक्शन की अनुमोदित लेबलिंग में इतना ढीला था प्रत्येक अन्य दवा - जिसमें एमआरएनए-आधारित ओनपैट्रो भी शामिल है - उस जानकारी को निर्दिष्ट करती है. ऐतिहासिक रूप से, एफडीए नियामक निर्णय (जैसे कि उत्पाद लेबलिंग में कौन सी जानकारी शामिल करनी है) प्राथमिकता पर आधारित होते हैं, और कोविड एमआरएनए शॉट्स एफडीए की ऐतिहासिक और कानूनी प्राथमिकता से एक स्पष्ट विचलन थे। वह उल्लेखनीय डेटा अनुपस्थिति और स्पष्टता की कमी पुराने दिनों की याद दिलाती है मॉर्ले का लीवर और किडनी सौहार्दपूर्ण 1800 के अंत में. अंतर यह है: तब, एफडीए अस्तित्व में नहीं था, लेकिन आज ~20,000 कर्मचारियों वाला एक एफडीए है, जिनमें से कम से कम कुछ लोग स्पष्ट रूप से मानते थे कि यह लेबल पारदर्शी और "सच्चा" था।
किसी अज्ञात/अस्पष्ट/अस्पष्ट घटक को बताना, जिसे कोई भी सटीक रूप से निर्धारित नहीं कर सकता है, 1906 के शुद्ध खाद्य और औषधि अधिनियम के कानून निर्माताओं का इरादा नहीं था जब उन्होंने "सच्चे लेबलिंग" पर एफडीए नियमों को निर्दिष्ट किया था। उससे अलग: तथ्य यह है कि विभिन्न निर्माताओं से खुराक प्रति मात्रा दोगुनी हो जाती है (30mcg/0.3mL vs 100mcg/0.5mL) का अर्थ है कि ये एमआरएनए अनुक्रम न्यूक्लियोटाइड लंबाई में काफी भिन्न प्रतीत होते हैं, और बदले में, होंगे अधिक और विभिन्न एलएनपी और अनुलग्नक. क्या इसका मतलब यह है कि स्पाइक प्रोटीन को ट्रांसक्राइब करने के लिए उपयोग किए जाने वाले एमआरएनए अनुक्रम विभिन्न निर्माताओं की तुलना में लगभग दोगुने आकार (10mcg/0.1mL बनाम 20mcg/0.1mL) के हैं, या न्यूक्लियोटाइड लंबाई के अंतर में कुछ और योगदान दे रहा है?
आम आदमी के लिए अभी भी इस बिंदु तक पढ़ना (वैसे, बधाई): विस्तृत लेबलिंग जानकारी का अभाव मोटे तौर पर बिक्री के लिए एक घर का विज्ञापन करने जैसा हो सकता है, जिसमें कहा गया है कि यह सीमेंट स्लैब पर लकड़ी और ईंटों से बना है - लेकिन दिखाया नहीं जा रहा है घर की कोई भी तस्वीर, (उदाहरण के लिए अनुक्रम) और इसके वर्ग फ़ुटेज (उदाहरण के लिए आणविक भार) को साझा नहीं करना। किसी भी मामले में, जानकारी की कमी अपर्याप्त है और पारंपरिक मानकों का विचलन है।
अन्य एफडीए-अनुमोदित दवा - जिसमें अन्य एमआरएनए दवाएं भी शामिल हैं - में उनके उत्पादों पर पूर्ण घटक का खुलासा शामिल है एक संरचनात्मक प्रतिनिधित्व और आणविक भार उनके उत्पाद के बारे में ताकि लोगों को ठीक-ठीक पता चले कि उन्हें क्या मिल रहा है।
यह सच है: आप जिस भी दवा के बारे में सोच सकते हैं उसे देखें ड्रग्स.कॉम डेटाबेस और ध्यान दें कि कैसे सभी लेबल संरचना और/या आणविक भार प्रदान करते हैं। सबूत है कि कोविड एमआरएनए शॉट्स एक हैं विशिष्ट ऐतिहासिक FDA अनुमोदन अभ्यास और "सच्चा लेबल" नियम का अपवाद।
2023 डेनिश अध्ययन विवरण एमआरएनए कोविड-19 एमआरएनए इंजेक्शन के बैचों के बीच महत्वपूर्ण नैदानिक परिवर्तनशीलता:
यहां तक कि संभावित रूप से अमान्य "मेल-इन" परीक्षण सत्यापन पर भी कोई पारदर्शिता नहीं होने से निर्माताओं को एफडीए की देखरेख के एक और महत्वपूर्ण हिस्से पर छूट मिल गई है: एमआरएनए शॉट्स के लॉट/बैच विविधताओं पर संभावित नैदानिक अभिव्यक्तियाँ। एक पूर्वव्यापी डेनिश सुरक्षा अध्ययन इससे पहले 2023 में प्रकाशित फाइजर-बायोएनटेक बीएनटी162बी2 एमआरएनए इंजेक्शन से प्रतिकूल घटना रिपोर्टों का एक अत्यधिक विचलित पैटर्न डेनिश डीकेएमए प्रतिकूल घटना रिपोर्टिंग प्रणाली के साथ सहसंबद्ध था।
निम्नलिखित लाइन ग्राफ में, अलग-अलग रंग के बिंदु फाइजर-बायोएनटेक के एमआरएनए इंजेक्शन के विभिन्न बैचों का प्रतिनिधित्व करते हैं। इसने बैचों को तीन अलग-अलग श्रेणियों में अलग कर दिया; रिपोर्ट किए गए प्रतिकूल घटना समूहों (क्रमशः नीले, हरे और पीले प्लॉट) की उच्च-निम्न-से (लगभग) अनुपस्थित संख्या।
दूसरे शब्दों में: एक ही निर्माता के अनुमानित रूप से "समकक्ष" उत्पादों में बैच के अनुसार प्रतिकूल घटनाओं की बेतहाशा भिन्न घटनाएं होती हैं, जिनमें से प्रत्येक बैच सैकड़ों हजारों एमआरएनए इंजेक्शन का प्रतिनिधित्व करता है।
जब संगत रैखिक प्रतिगमन रेखाएँ जोड़ी गईं, तो एक विशेष पैटर्न उभरा:
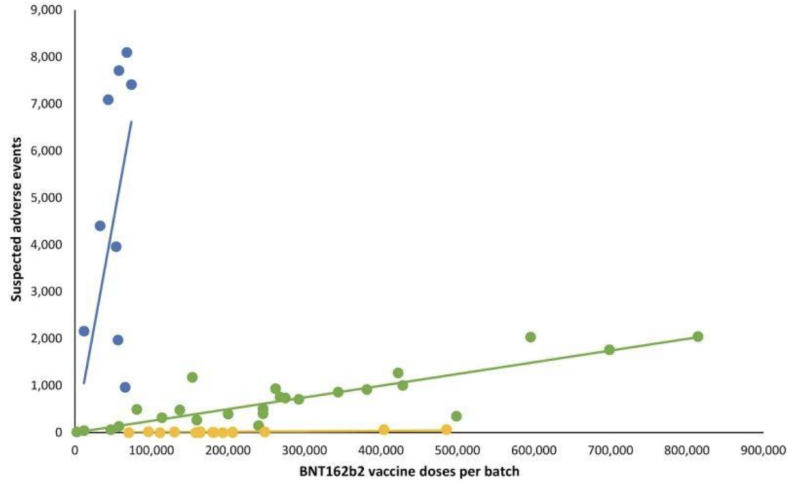
कोविड-19 एमआरएनए बैचों के बीच उल्लेखनीय प्रतिकूल घटनाओं की असमानता के बारे में महत्वपूर्ण प्रश्नों में शामिल हैं:
- क्या प्रतिकूल घटना भिन्नताएं एमआरएनए अनुक्रमों में गुणात्मक या मात्रात्मक भिन्नताओं या बैचों के बीच एमआरएनए स्ट्रैंड की संख्या के कारण हो सकती हैं?
- क्या प्रतिकूल घटना भिन्नता बैचों के बीच एलएनपी के आकार/आकृति या मात्रा में गुणात्मक या मात्रात्मक भिन्नता के कारण हो सकती है? कौन से परीक्षण किए गए हैं विभिन्न एलएनपी की सुरक्षा सुनिश्चित करें एमआरएनए इंजेक्शन में उपयोग किया जाता है?
- क्या वे बैच जो पीले बनाम हरे बनाम नीले डेटा बिंदुओं से मेल खाते थे, वे किसी तरह गुणात्मक या मात्रात्मक रूप से भिन्न थे?
- क्या निर्माण के बाद प्रबंधन सुविधा (या आपूर्ति श्रृंखला के साथ कहीं और) में भंडारण/हैंडलिंग से समझौता किया गया था, जिससे उत्पाद परिवर्तनशीलता हुई?
- विशेष विनिर्माण सुविधा/विनिर्माण के प्रभारी शिफ्ट प्रमुख से उत्पन्न होने वाले इस और अन्य उत्पादों की सिग्मा/त्रुटि दर क्या है?
- क्या बैच के आधार पर, कोविड एमआरएनए उत्पादों की सामग्री भारत या चीन बनाम कहीं और से ली गई थी?
- शुरुआत से आज तक "मेल द्वारा भेजे जाने" की तुलना में एफडीए निरीक्षक द्वारा व्यक्तिगत संग्रह के माध्यम से कोविड एमआरएनए उत्पादों के कितने प्रतिशत बैचों का परीक्षण किया गया था? क्या प्रत्येक बैच का परीक्षण केवल इन दो संग्रह विधियों में से किसी एक का उपयोग करके किया गया था?
- क्या FDA ने डेनिश DKMA प्रतिकूल घटना रिपोर्टिंग सिस्टम लॉट पर रिलीज़ परीक्षण सत्यापन किया था? यदि हाँ, तो FDA उन विशेष परीक्षण परिणामों को जारी क्यों नहीं कर रहा है? यदि नहीं, तो परीक्षण क्यों नहीं किया गया?
- क्या एलएनपी और/या एमआरएनए अनुक्रमों को लगातार विश्वसनीय और बिना संदूषण के उत्पादित करने में कोई बुनियादी समस्या है?
डेनिश अध्ययन के नतीजे और प्रतिकूल घटनाओं के बारे में उपरोक्त प्रश्नों को संबोधित करना *शुरू* किया जा सकता है, लेकिन एफडीए द्वारा स्वतंत्र रूप से उनके रिलीज परीक्षण निष्कर्षों के परिणामों को साझा किए बिना नहीं। जैसा कि यह है, सर्वव्यापी एफडीए (बी)(4) कटौती के कारण, कोई भी कोविड एमआरएनए शॉट्स के परीक्षण के लिए मान्य पद्धति को नहीं जानता है or वास्तव में डेनिश अध्ययन में कौन से लॉट का परीक्षण किया गया था या नहीं किया गया था or उन बैच परीक्षणों के परिणाम।
...तो फिर, भले ही एफडीए ने उन बैच परीक्षण परिणामों को जारी करने का फैसला किया हो, उपभोक्ताओं को कैसे पता चलेगा कि वे परिणाम निर्दिष्ट बैचों के प्रतिनिधि हैं, क्योंकि निर्माता स्वयं चयन कर रहे हैं कि कौन से नमूने "मेल में" भेजे जाएं?
घटक पारदर्शिता प्रदान नहीं करना और उचित नमूना पद्धति के माध्यम से गुणवत्ता सुनिश्चित करना एफडीए की एक मौलिक और बुनियादी आवश्यकता है। वास्तव में, यह FDA के गठन का प्राथमिक कारण था! जब हमारे फार्मास्यूटिकल्स की बात आती है तो क्या अमेरिकी बेहतर पारदर्शिता, निरीक्षण और "सच्चे लेबलिंग" कानूनों के हकदार नहीं हैं - खासकर जब से वे कानून 100 साल पहले बनाए गए थे?
ए के तहत प्रकाशित क्रिएटिव कॉमन्स एट्रिब्यूशन 4.0 इंटरनेशनल लाइसेंस
पुनर्मुद्रण के लिए, कृपया कैनोनिकल लिंक को मूल पर वापस सेट करें ब्राउनस्टोन संस्थान आलेख एवं लेखक.








